![]()
JOURNAL OF NEPHROLOGY Vol. 10 no. 5 - 1997 / pp 253-257
Hemolytic uremic syndrome/thrombotic thrombocytopenic purpura (HUS/TTP) complicating adult Still's disease: remission induced with intravenous immunoglobulin G
Jonathan R. Diamond - Division of Nephrology Milton Hershey Medical Center and the Pennsylvania State University College of Medicine, Hershey, PA, USA
ABSTRACT: Coexistence of hemolytic uremic syndrome/thrombotic thrombocytopenic purpura (HUS/TTP) and adult Still's disease is extremely rare. We describe the case history of a 22-year-old young man who presented with evidence of a thrombotic microangiopathy complicated by dialysis-dependent renal failure, encephalopathy, and an ischemic retinopathy. The most important and novel feature of this case was the dramatic and sustained clinical remission of the TMA induced by intravenous immunoglobulin (IVIg) after failure of plasmapheresis and glucocorticoids to do so.
KEY WORDS: Hemolytic uremic syndrome, Thrombotic thrombocytopenic purpura, Intravenous immunoglobulin G, Plasmapheresis, Cryosupernatant, Fresh frozen plasma, Renal failure, Dialysis
Introduction
Hemolytic uremic syndrome (HUS) is a disorder that mainly affects infants and small children, although adults may also acquire the disease. HUS is comprised of a non-immune hemolytic anemia, renal failure, and thrombocytopenia. The vasculopathy of HUS and of thrombotic thrombocytopenic purpura (TTP) are virtually identical and investigators have argued that it may represent a continuum of disease and, perhaps, the term thrombotic microangiopathy be utilized (1). We present a case of a young man who developed a thrombotic microangiopathy (TMA), namely idiopathic HUS/TTP, complicating adult Still's disease that chiefly manifested itself as dialysis-dependent renal failure, non-immune hemolytic anemia, thrombocytopenia, encephalopathy, and an ischemic retinopathy.
Still's disease was originally described by George Frederick Still (2) and is a variant of juvenile rheumatoid arthritis with significant systemic features including arthritis, fever, rash, lymphoid hypertrophy, leukocytosis, pericarditis, hepatomegaly, anemia, and thrombocytosis. The most important and novel feature of this case was the dramatic and sustained clinical remission of the TMA induced by intravenous immunoglobulin (IVIg) after failure of plasmapheresis and glucocorticoids to do so.
Case Report
T.R. is a 22 year-old white male referred to our institution for 3acute renal failure. Four months prior to admission, he developed fever, an evanescent macular rash, arthralgias, and lymphopenia. His serum ferritin was 4500 ng/ml and a clinical diagnosis of adult Still's disease was made. He was treated with a nonsteroidal anti-inflammatory agent with improvement of his symptoms. One month prior to admission, he was admitted to an outlying hospital with pleurisy and was started on oral prednisone (10 mg/d) with improvement. On the day prior to admission, the patient presented to his local physician with intractable nausea and vomiting, myalgias, blurred vision and was noted to be in acute renal failure with a blood urea nitrogen of 160 mg/dl, a serum creatinine of 10.7 mg/dl, and an elevated anion gap metabolic acidosis. His renal parameters during the previous month's hospitalization were all normal. At no time during his four-month illness did the patient have a diarrheal component or upper respiratory tract prodrome.
On physical examination, the blood pressure was 110/60 mmHg. with a pulse of 80 beats/minute. He was in no respiratory distress with a respiratory rate of 14/minute and he was afebrile. On fundoscopic examination, a diagnosis of Purtscher's-like retinopathy and marked capillary nonperfusion in both eyes was made and was felt to be ischemic in nature. Chest examination revealed bilateral pleural effusions. There was a 2-component pericardial friction rub. The liver was enlarged with the edge palpable 2-3 fingerbreadths below the right costal margin. There was no splenomegaly. Neurologically, there was no asterixis and he was without lateralizing signs or evidence of encephalopathy.
A urinalysis exhibited 4+ albuminuria with many darkly-pigmented granular and tubular epithelial cell casts as well as calcium oxalate (dihydrate) and uric acid crystals. The sediment was consistent with acute tubular necrosis. A random urine protein to creatinine ratio estimated the 24 hr protein excretion to be ~ 4 grams per day. The complete blood count was white blood count 15.9 k/ul (differential: 95% polymorphonuclear leukocytes, 4% lymphocytes, 1% immature granulocytes), hemoglobin 10.2 g/dl, hematocrit (Hct) 29.8%, platelet count 134 k. The peripheral blood smear exhibited polychromasia, many schistocytes, and moderate basophilic stippling. An erythrocyte sedimentation rate was 37 mm/hr. Serum ferritin was 5545 ng/ml, fibrinogen 242 mg/dl, prothrombin time 14 secs (1.3 INR), partial thromboplastin time 28 secs, D-dimer 0.5 ug/ml. Other serum chemistries included calcium 8.0 mg/dl, uric acid 12.9 mg/dl, phosphorus 12.8 mg/dl, magnesium 2.5 mg/dl, total bilirubin 1.2 mg/dl, albumin 3.1 g/dl. A lactic dehydrogenase (LDH) level was elevated at 1800 IU/L on the day of admission but promptly rose to 3281 IU/L on the second hospital day.
A percutaneous kidney biopsy was performed on the second hospital day and demonstrated plugging of many of the glomerular capillary loops with fine granular material and fragmented red blood cells (fig. 1). The glomeruli showed only a minimal increase in cellularity. Thrombi were noted in blood vessels adjacent to the glomerular tuft. Extensive damage to the renal cortical tubules was observed with marked tubular dilatation as well as granular material filling a number of lumina. Immunofluorescence was noteworthy for moderate staining for fibrin in the majority of glomeruli examined (fig. 2). On electron microscopy, most capillary loops were occluded by either monocytes or electron dense material consistent with fibrin admixed with platelets (fig. 3). Epithelial foot processes were diffusely fused. The changes observed were consistent with a microangiopathic glomerulopathy. The tubular damage was putatively ischemic from obstruction to glomerular blood flow.
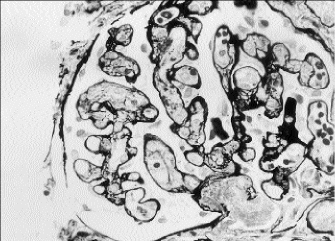
Fig. 1 - Photomicrograph of a representative glomerulus demonstrating many capillary loops with fine granular material and fragmented red blood cells within them. Some patent capillary loops are also present (methenamine silver, X400).
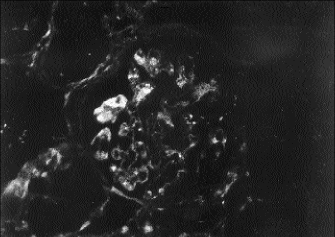
Fig. 2 - Immunofluorescent microscopy labeling for fibrin. As is evident, there was a moderate staining intensity for this clotting factor within the capillary loops of this representative glomerulus (X 300).
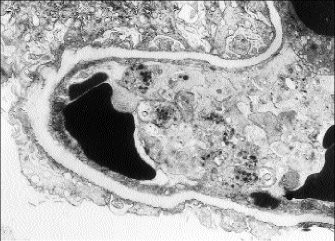
Fig. 3 - A representative glomerular capillary loop obstructed with electron dense material consistent with fibrin admixed with platelets. Epithelial foot processes are diffusely fused.
A diagnosis of hemolytic uremic syndrome/thrombotic thrombocytopenic purpura (HUS/TTP) complicating adult Still's disease was made. Within 24 hrs of kidney biopsy, the patient exhibited waxing and waning mental status without lateralizing signs. A diagnosis of encephalopathy, which did not improve with hemodialysis, was made. On hospital day 2 (fig. 4), plasmapheresis with fresh frozen plasma replacement was commenced and was continued daily through the 14th hospital day. For the 7th apheresis (and for all subsequent ones), cryosupernatant was utilized rather than fresh frozen plasma. Despite an initial response to the daily plasmapheresis, as evidence by a decline in the serum LDH level to 1700 IU/L on the 5th hospital day, on-going intravascular hemolysis was detected by a dramatic fall in Hct. and platelet count coupled with a near-doubling of the serum LDH on the 6th hospital day. The patient was intermittently (days 7, 9, and 11) transfused with a total of 6 units packed red blood cells and was begun on intravenous methylprednisolone 120 mg/day on 7th hospital day. The glucocorticoid dose was decreased to 60 mg/day on the 10th hospital day, but due to a marked rise in serum LDH level to 6054 IU/L on the 13th hospital day, the methylprednisolone dose was increased to 120 mg/day. Due to a failure to induce a remission of the HUS/TTP disorder with plasmapheresis with either fresh frozen plasma or cryosupernatant as well as methylprednisolone, intravenous immunoglobulin G (IVIg, Baxter Healthcare Corp.) 1 gm/kg/day for 3 days was commenced on the 13th hospital day. Within 12 hours of the administration of IVIg, the serum LDH level promptly declined to 1802 IU/L and did not increase any further. Without further packed red blood cell transfusion, the Hct. rose to a maximum of 32.7% on the 16th hospital day. The platelet count did not change appreciably until after discharge, when it increased to and was sustained at greater than 300k. No plasmapheresis was required after the second dose of IVIg. The glucocorticoid dose was rapidly tapered from 120 mg/day to 30 mg/day at the time of discharge (hospital day #21). The patient's renal course was consistent with severe oliguric acute tubular necrosis. There was a gradual increase in the daily urine volume and after 16 consecutive daily hemodialysis treatments, renal replacement therapy was discontinued on hospital day #17. At discharge the serum creatinine had declined to 1.7 mg/dl and normalized to 1.0 mg/dl as an outpatient.
As an outpatient, the patient received IVIg (1 gm/kg) every 2 weeks for 1 month and then at monthly intervals for an additional 2 months. This has resulted in a Hct. of 35.7%, a platelet count of 317k, a serum LDH of 553 IU/L, and a serum creatinine of 1.0 mg/dl at day 62 after his presentation to this hospital with acute renal failure.
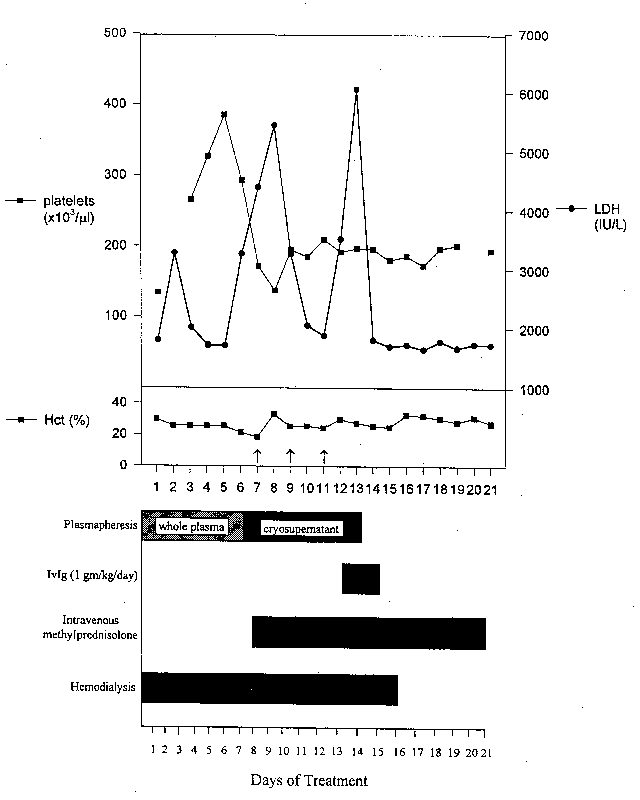
fig. 4 - Summary of the clinical course. The platelet count and LDH values are in the upper panel; the hematocrit (Hct.) is located in the middle panel; and the treatment modalities are presented as horizontal histogram bars in the lower panel. The breaks in the platelet count curve in the upper panel reflect an absence of data for this parameter on the second and twentieth days of treatment. There was a prompt and lasting response to IVIg (1 gm/kg/day), which was administered on days 13-15, as evidenced by a dramatic and sustained fall in the serum LDH level. The arrows in the middle panel show when 2 units of packed red blood cells were transfused on each of the three indicated days.
Discussion
Thrombotic microangiopathy complicating adult Still's disease has rarely been reported. Boki et al (3) have reported two patients with adult Still's disease who also had co-existing TTP. Bray et al (4) identified one patient and reviewed 15 other reported cases of disseminated intravascular coagulation complicating adult Still's disease. To our knowledge no case of dialysis-dependent HUS/TTP has been reported complicating adult Still's disease. The coexistence of two idiopathic disorders is very unusual and may suggest a similar pathogenesis involving endothelial cell dysfunction with coagulation abnormalities.
Atypical forms of adult HUS require more therapy than supportive care. Plasma infusion or exchange is the treatment of choice in adult and relapsing forms of HUS and when there are neurological signs (5, 6), as there were with this case. Substitution of the cryosupernatant fraction of plasma, devoid of the largest plasma von Willebrand factor (vWf) multimers, for whole plasma during plasmapheresis has also been reported to be successful in patients who did not respond to whole plasma infusion (5, 7). The patient reported, although initially responsive to both whole plasma and cryosupernatant infusions with plasmapheresis, ultimately failed both treatment modalities.
Other treatment modalities for HUS/TTP include anticoagulation, antiplatelet agents, prostaglandin (PGI2) infusion, high-dose corticosteroids, splenectomy, and vincristine. Of these modalities, only intravenous methylprednisolone was utilized in our patient with limited, if any, success. Raniele et al (8) have reported the dramatic efficacy of intravenous immunoglobulin G (IVIg) in a patient with acute TTP when other treatment strategies had been ineffective. As with their case (8), our patient exhibited a rapid (i.e., within 12-24 hrs) and sustained response to IVIg (1 gm/kg/day for 3 days). It has been speculated (8) that the administration of IgG may be effective by neutralizing cytotoxic or platelet agglutinating factors.
Although IVIg has the risks of anaphylaxis in IgA-deficient patients (9), the possibility of osmotic nephrosis (10), and infections secondary to inhibition of Fc receptor-mediated immune clearance (11) as well as the financial cost, this mode of therapy may be life-saving and induce remission of HUS-TTP when other costly and potentially noxious treatment options fail.
In summary, we report the
dramatic response of a patient with HUS/TTP complicating adult Still's disease
to IVIg. The case report anectdotally suggests that this form of treatment
should be studied prospectively to address its more frequent and earlier
utilization in HUS/TTP.
Reprint requests to: Jonathan R. Diamond, M.D. - Division
of Nephrology Milton S. Hershey Medical Center P.O. Box 850
Hershey, PA 17033, U.S.A.
References
1. Remuzzi G, Ruggenenti P. The hemolytic uremic syndrome. Kidney Int 1995; 47: 2-19.
2. Still GF. On a form of chronic joint disease in children. Med Chir Trans 1897; 80: 47-9.
3. Boki KA, Tsirantonaki MJ, Markakis K, Moutosoupoulos KM. Thrombotic thrombocytopenic purpura in adult Still's disease. J Rheumatol 1996; 23: 385-7.
4. Bray VJ, Singleton JD. Disseminated intravascular coagulation in Still's disease. Sem Arthrit Rheumat 1994; 24: 222-9.
5. Rugenenti P, Galbusera M, Plata CR, Bellavita P, Remuzzi G. Thrombotic thrombocytopenic purpura: Evidence that infusion rather than removal of plasma induces remission of the disease. Am J Kidney Dis 1993; 21: 314-8.
6. Sheeth KJ, Leichter HE, Gill HC, Baumgardt A. Reversal of central nervous system involvement in hemolytic uremic syndrome by use of plasma exhanges. Clin Pediatr 1987; 26: 651-6.
7. Byrnes JJ, Moake JL, Klug P, Perlman P. Effectiveness of the cryosupernatant fraction of plasma in the treatment of refractory thrombotic thrombocytopenic purpura. Am J Hematol 1990; 34: 169-74.
8. Raniele DP, Opsahi JA, Kjellstrand CM. Should intravenous immunoglobulin G be first-line tratment for acute thrombotic thromobcytopenic purpura?: Case report and review of the literature. Am J Kid Dis 1991; 18: 264-8.
9. Berkman SA, Lee ML, Gale RP. Clinical uses of intravenous immunoglobulins. Ann Intern Med 1988; 39: 302-12.
10. Ahsan N, Wiegand LA, Abendroth CS, Manning EC. Acute renal failure following immunoglobulin therapy. Am J Nephrol 1996; 16: 532-6.
11. Cross AS, Siegl G, Byrne WR. Intravenous immune globulins impairs anti-bacterial defenses of a cyclophosphamide-treated host. Clin Exp Immunol 1989; 76: 159-64.
Received: April 15, 1997 Accepted: May 5, 1997