BMJ 1997;314:583 (22 February)
![]()
Clinical review
Grand Rounds--University Hospital of Wales, Cardiff: Pyrexia of unknown origin
The difficulty of establishing a diagnosis
Case presented by: Richard H
Evans, registrar in medicine and infectious diseases
Chairman: R E Mansel, professor of surgery
Discussion group: L K Borysiewicz, professor of medicine, B D Williams, professor
of rheumatology
Department of medicine, University hospital of Wales, Heath park, Cardiff Cf4 4XW
Pyrexia of unknown origin is a common problem in medical practice and encompasses a broad spectrum of diagnostic possibilities. The clinical phenomenon of pyrexia of unknown origin was defined in 1961 as an illness persisting for three weeks or longer, with episodes of fever greater than 38.3°C and with no diagnosis reached after one week of hospital investigation.1 This case highlights the difficulty that can be encountered in establishing a diagnosis in a patient presenting with pyrexia of unknown origin.
Case history
A 34 year old man developed a mild sore throat on 29 October 1995.
This became more severe during the following week, when he consulted
his general practitioner and was given a seven day course of
penicillin V. The patient discontinued the antibiotics after two days
because of nausea. During the subsequent fortnight he developed
myalgia and arthralgia, followed by drenching night sweats and
rigors.
After four weeks he was unable to get out of bed without help because of the severity of the myalgia and arthralgia, and he had lost over 10 kg. He was admitted to his district general hospital in November 1995. He appeared pale and unwell with a fever of 40°C. He complained of weakness and intense myalgia of his shoulder and pelvic girdle muscles, arthralgia of his left knee and right elbow, and left sided pleuritic chest pain. He had no medical history of note and no history of recent foreign travel. Physical examination was unremarkable: there was no palpable lymphadenopathy; no organomegaly; and no stigmata of infective endocarditis.
Laboratory data on admission
showed a blood haemoglobin concentration of 81 g/l with normochromic
normocytic indices, a raised leucocyte count of 21.85x109/l
with a neutrophil count of 19.65x109/l and a raised
platelet count of 648x109/l. His erythrocyte sedimentation rate
and C reactive protein were both raised, at 124 mm in the first hour
and 276 mg/l (normal 0-6 mg/l) respectively. Renal function was
normal, but liver function tests were raised: aspartate aminotransferase 58
IU/l (5-45 IU/l); alkaline phosphatase 276 IU/l (30-115 IU/l); ![]() -glutamyltransferase
174 IU/l (5-48 IU/l); bilirubin 6 µmol/l (1-17 mol/l); albumin 28
g/l (35-50 g/l). Anti-streptolysin O titres were normal. The chest
radiograph was normal.
-glutamyltransferase
174 IU/l (5-48 IU/l); bilirubin 6 µmol/l (1-17 mol/l); albumin 28
g/l (35-50 g/l). Anti-streptolysin O titres were normal. The chest
radiograph was normal.
His fevers, myalgia, and arthralgia had not settled after one week in hospital, and he was started empirically on intravenous benzylpenicillin. Four days later he developed a mildly pruritic maculopapular rash over his trunk, arms, and groin. The rash was attributed to the penicillin–which was stopped–but the rash was still present one week later and seemed to be getting worse.
Serological tests failed to show any evidence of a bacterial or viral infection. Blood and urine cultures were negative. Antinuclear antibody, rheumatoid factor, anti-double stranded DNA antibodies, and anti-neutrophil cytoplasmic antibody concentrations were negative. Serum angiotensin converting enzyme concentrations were normal. A bone marrow aspirate showed a hypercellular marrow consistent with reactive change but without any evidence of lymphomatous or other malignant infiltration. The trephine biopsy was normal. A Ziehl-Neelsen stain of the bone marrow was negative for acid-alcohol fast bacilli, and a Mantoux test was negative. He developed a left knee effusion, an aspirate of which showed numerous polymorphs but no organisms or crystals, and culture was negative. Plain radiographs of the affected joints were normal, as were an ultrasound scan of his abdomen, a computed tomogram of his thorax, abdomen, and pelvis, and an isotope lung scan. A transthoracic echocardiogram showed normal heart function with no evidence of infective endocarditis. An indium-111 labelled white cell scan failed to show any focus of infection or inflammation.
Seven weeks after the initial onset of his illness he was transferred to the infectious diseases unit at this hospital for further assessment. He had lost over 22 kg and continued to complain of generalised myalgia and arthralgia of his wrists, left knee, and right shoulder. The left knee effusion was still clinically apparent, and the movement of his right shoulder was restricted. There was evidence of synovitis of both carpometacarpal joints with restriction to extension. The maculopapular rash over the trunk, upper limbs, and groin became more florid during fevers, which were occurring regularly at 10 pm each night and were preceded by intense rigors and sweats. Physical examination was otherwise normal. In addition to the previously documented laboratory abnormalities, he was found to have a greatly raised serum ferritin concentration of 12 000 µg/l (normal 15-300 µg/l). Transoesophageal echocardiography, performed to exclude endocarditis, was normal.
Diagnosis
The differential diagnosis of fever and arthritis is wide (box), and
most causes had been excluded by the exhaustive investigations. We
thought that the pattern of his illness was most suggestive of acute
onset connective tissue disease. In this clinical context, and with
the benefit of the widespread negative investigations, we were able
to secure the diagnosis of adult Still's disease.
Differential
diagnosis of fever and arthritis2
Direct (bacterial, fungal) Indirect (bacterial: acute rheumatic fever, reactive arthritis; viral: hepatitis B)
Monosodium urate (gout) Calcium pyrophosphate (pseudogout)
Systemic lupus erythematosus Mixed connective tissue disease Necrotising vasculitis and other vasculopathies Adult onset Still's disease Rheumatoid arthritis Polymyalgia rheumatica
|
Treatment
The patient was treated with pulses of 1 g intravenous methylprednisolone on
three consecutive days. His symptoms improved dramatically within 12
hours of starting treatment: his fever settled and never returned
(fig 1); the myalgias and joint pains disappeared, and
he was able to get out of bed without help. He was subsequently started on
oral prednisolone 30 mg and azathioprine 150 mg.
|
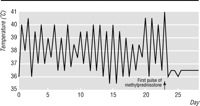
Four weeks after discharge from hospital the patient remained asymptomatic. His erythrocyte sedimentation rate had fallen to 50 mm in the first hour and the serum ferritin concentration to 400 µg/l. His only complaint was of some persistent restriction in wrist extension.
Comment
The diagnostic spectrum of pyrexia of unknown origin has changed since
its original definition over 30 years ago. The incidence of rheumatic
fever has declined; infective endocarditis is diagnosed more readily
by the routine use of blood cultures and echocardiography; advances
in imaging techniques now allow earlier diagnosis of solid tumours;
and the general health of the population has improved. As the number
of cases of pyrexia of unknown origin due to infectious diseases has
declined there has been a corresponding increase in the number of
cases due to multisystem diseases–a significant proportion of these
patients have adult Still's disease.3 4
5
Still's disease, the systemic form of juvenile chronic arthritis, was first described in 1897,6 but Still's disease in the adult was not recognised until 1971.7 Adult Still's disease characteristically affects young adults, with 75% of patients first presenting between the ages of 16 and 35 years, although several cases have been reported with disease onset after the age of 60 years.8 9 Still's disease affects men and women about equally. Although data on incidence and prevalence vary, the disease is not thought to occur in more than 1 case per 100 000 population.10
The cause of adult Still's disease is unclear. There are no consistent associations with HLA antigens,2 11 2 and although numerous infectious agents have been implicated as causative agents13 14 15 16 17 the precise role of micro-organisms has yet to be established. It is likely that immune, infectious, and environmental factors may have a role in a genetically predisposed individual.
The diagnosis of adult Still's disease is based entirely on clinical findings (box). Since no reliable serological markers or other diagnostic tests exist, recognition of the classic features of adult Still's disease remains the key to the diagnosis. Fever, the most constant feature of the disease, is high and spiking–the spike typically occurring in the early evening and falling to normal at least once during a 24 hour period. Most patients have pronounced myalgias, often accompanied by polyarthralgias around large joints, commonly the knees, fingers, and wrists. The rash, often said to be the most useful feature for diagnosis, is salmon-pink, macular or maculopapular, and evanescent. It is often pruritic and is frequently misinterpreted as a drug allergy.19 A sore throat is also said to be characteristic of the disease as no pathogens are isolated, anti-streptolysin O titres remain normal, and because a sore throat is rarely a feature of other connective tissue diseases.20 Other common features of the condition include lymphadenopathy, splenomegaly, pericarditis, and pleuritis.2 18 21 The early clinical presentation of this condition, however, is extremely variable, making the diagnosis difficult. The classic triad of fever, arthritis, and rash occurs in fewer than 50% of cases at the time of presentation.18
| Principal diagnostic
criteria for adult Still's disease18
Major criteria Fever of 39°C Arthralgia Typical rash Neutrophil leucocytosis Minor criteria Sore throat Lymphadenopathy or splenomegaly, or both Liver dysfunction Negative rheumatoid factor, and negative antinuclear antibody Exclusions Infections Malignancies Rheumatic diseases Suggested criteria for the diagnosis of adult Still's disease, requires five or more criteria, including two or more major criteria |
Laboratory investigations serve to exclude other diagnostic possibilities. Almost all patients with adult Still's disease have a high erythrocyte sedimentation rate and a neutrophil leucocytosis. Many patients have a normochromic normocytic anaemia and abnormal liver function tests. Tests for serum antinuclear antibody concentration and IgM rheumatoid factor are typically negative.2 18 2
Recent studies have shown a correlation between disease activity in adult Still's disease and extremely high concentrations of serum ferritin.21 22 23 Raised serum ferritin concentrations are frequently observed as part of the host response to systemic inflammation. Noticeably raised concentrations are seen in patients with haematological and germ cell malignancies, with acute liver necrosis, and in haemachromatosis, but concentrations rarely exceed 3000 µg/l. In contrast, serum ferritin concentrations above 10 000 µg/l have been noted in patients with adult Still's disease in the active phase of their illness and may therefore be useful not only in the diagnosis of the disease but also in monitoring disease activity.
Aspirin and other non-steroidal anti-inflammatory agents have been used successfully in the treatment of adult Still's disease, though a significant proportion of patients may need either systemic or oral corticosteroid treatment to control their disease. Pulsed intravenous methylprednisolone has been shown to be effective in treating evolving adult Still's disease and, when combined with low dose oral prednisolone, is also effective in preventing short term relapse.2 24 2
Discussion
REM: Were all of these investigations strictly necessary to obtain
the diagnosis?
LKB: Yes. The diagnosis is based on the clinical findings alone and relies heavily on exclusion of underlying infection or malignancy. Giving high doses of intravenous corticosteroids may prove catastrophic if the diagnosis is not secure. The patient's presentation was incomplete at first, and the investigations performed at the district general hospital were therefore entirely appropriate. Only with the benefit of these negative investigations could we be confident of the diagnosis.
LKB: Is the mechanism of production of these very high serum ferritin concentrations known?
RHE: Moderately raised concentrations probably arise from iron dependent regulation of ferritin synthesis. The greatly raised concentrations in adult Still's disease are likely to be due to a different mechanism: pronounced increases in ferritin synthesis have been observed in human hepatoma cells in response to interleukin 1ß, suggesting direct activation of ferritin mRNA translation by cytokines22; abnormalities in the glycosylation of ferritin have been shown in some patients with adult Still's disease, a process that may lead to impaired secretion and accumulation in the endoplasmic reticulum.23 It remains unclear, however, which one (or more) of these mechanisms is responsible for the pronounced hyperferritinaemia seen in adult Still's disease.
LKB: Have any other treatment options been tested?
BDW: The response of patients with adult Still's disease to treatment is difficult to evaluate, partly because the relative rarity of the disease does not lend itself to randomised controlled trials and partly because of the remittent nature of this disorder. High doses of salicylates or other non-steroidal agents have traditionally been used as first line treatment but are effective in only about 20% of patients.19 26 Some of the largest studies have shown that between 50% and 100% of patients with adult Still's disease need corticosteroids to control systemic disease.20 27 28 29 Unfortunately, even in those patients who respond to corticosteroids, joint destruction may progress when the systemic symptoms are adequately controlled, and the disease frequently relapses when the dose is reduced. Other agents have been used, but the evidence in favour of these agents is based either on small studies or on single case reports. Several immunosuppressive agents have been used with some success as steroid sparing agents, and remittive agents have been used to control joint symptoms. An initial trial with a non-steroidal agent may be appropriate, but corticosteroid treatment is needed to control the severe systemic symptoms in the most patients. Steroid sparing or remitting agents should be added to the treatment regimen as the steroids are withdrawn.
REM: What is the prognosis for patients with adult Still's disease?
BDW: The early reports of adult Still's disease suggested that it was a relatively benign condition. Recent studies have suggested a more ominous prognosis with recurrence of disease activity being characteristic, and an appreciable number of patients progressing to a chronic disease. Estimates on the proportion of patients who progress to this chronic disease course differ,20 27 20 but polyarticular onset, root joint involvement at presentation, or the presence of a typical Still's rash have been identified as prognostic markers associated with a chronic disease course and a prolonged time to remission.12 19 12 Despite the frequently chronic nature of this illness, disease flares in adult Still's disease are usually milder than the initial episode,19 and assessments of functional outcome have been quite favourable: about 80% of patients have a functional class of I or II, according to the American Rheumatism Association's classification. 12 29
| Acknowledgements |
|---|
We are grateful to Dr O M Gibby (consultant physician, Royal Gwent Hospital, Newport) for referring this patient to our unit, and for the patient's permission to publish his case.
| Notes |
|---|
The BMJ welcomes grand rounds from other hospitals.
| References |
|---|
- Petersdorf RG, Beeson PB. Fever of unexplained origin: report on 100 cases. Medicine 1961;40:1-30.
- Van de Putte LB, Wouters JG. Adult onset Still's disease. In: Sturrock RD, ed. Rheumatic manifestations of haematological disease. Clinical rheumatology international practice and research. Vol 5(2). London: Baillière Tindall, 1991:263-75.
- Petersdorf RG. Fever of unknown origin–an old friend revisited. Arch Intern Med 1992;152:21-2.
- Larson EB, Featherstone HJ, Petersdorf RG. Fever of undetermined origin: diagnosis and follow-up of 105 cases 1970-1980. Medicine 1982;61:269-92. [Medline]
- Kashiwagi H. Fever of unknown origin: a changing diagnostic spectrum. Intern Med 1994;2:65-6.
- Still GF. On a form of chronic joint disease in children. Med Chir Trans 1897;80:1-13 (reprinted Am J Dis Child 1978;132:195-200).
- Bywaters EG. Still's disease in the adult. Ann Rheum Dis 1971;30:121-33. [Medline]
- Steffe LA, Cooke CL. Still's disease in a 70-year-old woman. JAMA 1983;249:2062-3.
- Wouters J, Van Rijswijk M, Van de Putte L. Adult onset Still's disease in the elderly: a report of two cases. J Rheumatol 1985;12:791-3.
- Magadur-Joly G, Billaud E, Barrier JH, Pennec YL, Masson C, Renou P, et al. Epidemiology of adult Still's disease: estimate of the incidence by a retrospective study in west France. Ann Rheum Dis 1995;54:587-90. [Medline]
- Miller ML, Aaron S, Jackson J, Fraser P, Cairns L, Hoch S, et al. HLA gene frequencies in children and adults with systemic onset juvenile rheumatoid arthritis. Arthritis Rheum 1985;28:146-50.
- Wouters JM, Reekers P, van de Putte LB. Adult-onset Still's disease: disease course and HLA associations. Arthritis Rheum 1986;29:415-8. [Medline]
- Huang SH, De Coteau WE. Adult-onset Still's disease: an unusual presentation of rubella infection. Can Med Assoc J 1980;122:1275-6.
- Gordon SC, Lauter CB. Mumps arthritis: unusual presentation as adult Still's disease. Ann Intern Med 1982;97:45-7.
- Colebunders R, Stevens WJ, Vanagt E, Snoeck J. Adult Still's disease caused by Yersinia enterocolitica infection. Arch Intern Med 1984;144:1880-2.
- Hurst NP, Martynoga AG, Nuki G, Sewell JR, Mitchell A, Hughes GR. Coxsackie B infection and arthritis. BMJ 1983;286:605.
- Wouters JM, van der Veen J, van de Putte LB, de Rooy DJ. Adult onset Still's disease and viral infections. Ann Rheum Dis 1988;47:764-7.
- Yamaguchi M, Ohta A, Tsunematsu T, Kasukawa R, Mizushima Y, Kashiwagi H, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol 1992;19:424-30.
- Pouchot J, Sampalis JS, Beaudet F, Carette S, Décary F, Salusinsky-Sternbach M, et al. Adult Still's disease: manifestations, disease course, and outcome in 62 patients.Medicine 1991;70:118-36. [Medline]
- Larson EB. Adult Still's disease–evolution of a clinical syndrome and diagnosis, treatment, and follow-up of 17 patients. Medicine 1984;63:82-91. [Medline]
- Ohta A, Yamaguchi M, Kaneoka H, Nagayoshi T, Hiida M. Adult Still's disease: review of 228 cases from the literature. J Rheumatol 1987;14:1139-46. [Medline]
- Schwarz-Eywill M, Heilig B, Bauer H, Breitbart A, Pezzutto A. Evolution of serum ferritin as a marker for adult Still's disease activity. Ann Rheum Dis 1992;51:683-5. [Medline]
- Van Reeth C, Le Moel G, Lasne Y, Revenant M-C, Agneray J, Kahn M-F, et al. Serum ferritin and isoferritins are tools for diagnosis of active adult Still's disease. J Rheumatol 1994;21:890-5. [Medline]
- Bisagni-Faure A, Job-Deslandre C, Menkes C J. Intravenous methylprednisolone pulse therapy in Still's disease. J Rheumatol 1992;19:1487-8. [Medline]
- Khraishi M, Fam A. Treatment of fulminant adult Still's disease with intravenous pulse methylprednisolone therapy. J Rheumatol 1991;18:1088-90. [Medline]
- Aydintug AO, D'Cruz D, Cervera R, Khamashta MA, Hughes GRV. Low dose methotrexate treatment in adult Still's disease. J Rheumatol 1992;19:431-5. [Medline]
- Wouters JM, van de Putte LB. Adult onset Still's disease; clinical and laboratory features, treatment and progress of 45 cases. Q J Med 1986;61:1055-65. [Medline]
- Bujak JS, Aptekar RG, Decker JL, Wolff SM. Juvenile rheumatoid arthritis presenting in the adult as fever of unknown origin. Medicine 1973;52:431-44. [Medline]
- Cush JJ, Medsger TA, Christy WC, Herbert DC, Cooperstein LA. Adult onset Still's disease: clinical course and outcome. Arthritis Rheum 1987;30:186-94. [Medline]